Outline
What does dDocent do?
Quality Filtering
dDocent takes a minimalistic approach to quality filtering, using the program Trimmomatic. Low quality bases (below quality score of 20) are removed from the beginning and end of reads, and an additional sliding 5bp window that will trim bases when the average quality score drops below 10. Additionally, Illumina adapters are detected and removed. After quality filtering has been performed once, it does not need to be performed again. Files that have a .R1.fq and .R2.fq are the filtered FASTQ files. In theory, even more liberal parameters could be used because both read mapping and SNP calling incorporate base quality in evaluation.
De Novo Assembly
dDocent now has multiple different assembly algorithms for differing RADseq data types. The PE option is for Paired-End (PE) RADseq data that does not have a random shearing step (ddRAD and ezRAD). The RPE option is for PE data that comes from the original RAD method with a random shearing step. The OL algorithm is PE data where the read length is long compared to the targeted insert and substantial overlap between forward and reverse reads are expect. The SE algorithm can handle any type of RADseq methodology using single-end (SE) sequencing.
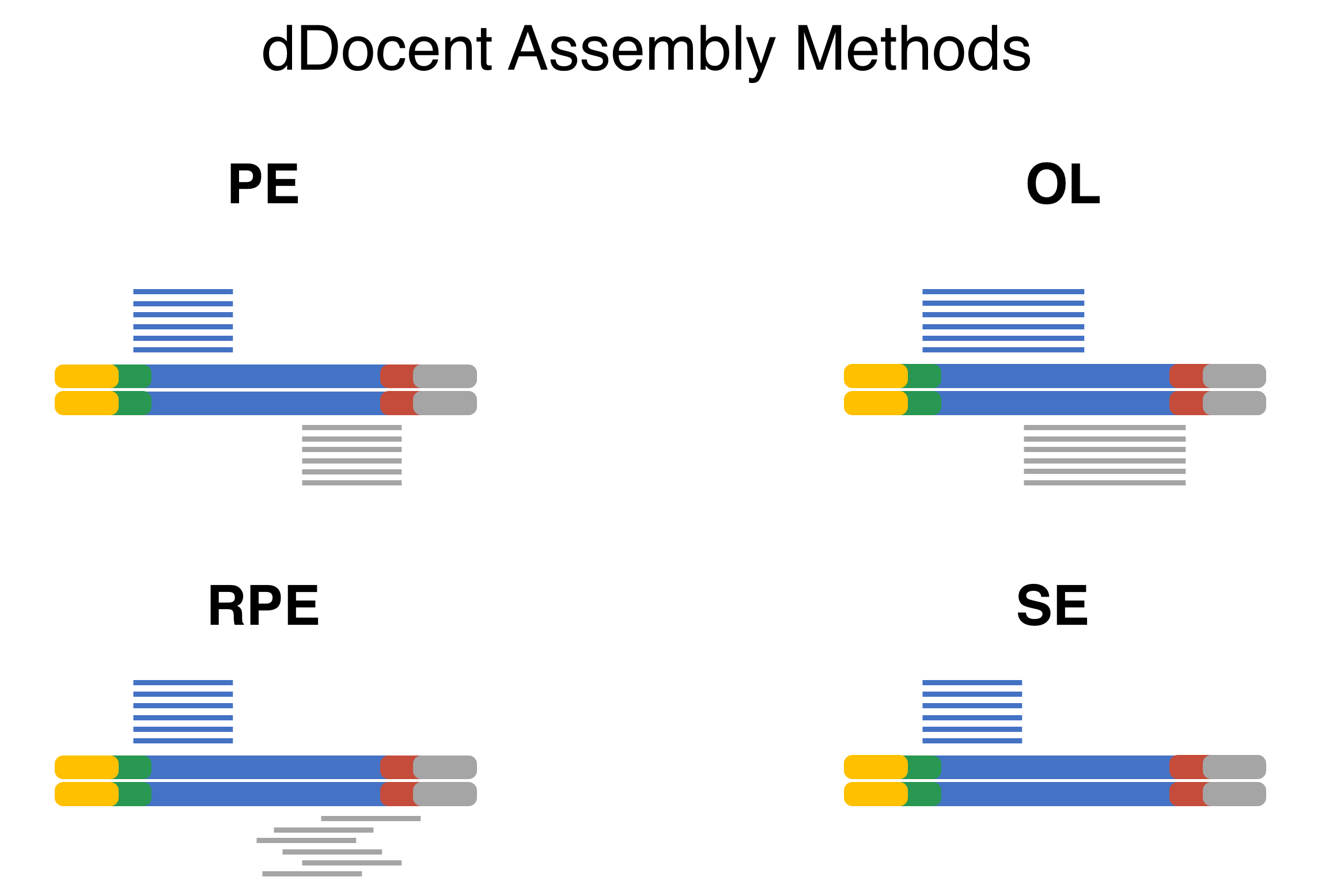
Data Reduction
For all assembly methods, dDocent uses a novel data reduction approach to help ensure accuracy. For PE methods, all reads are concatenated into single forward and reverse FASTA files, and for the SE method, only forward reads are used. From here, the complete set of unique reads (loci) are tabulated, along with the number of occurrences of each unique sequence. Reads that do not have a high number of occurrences are likely to be either sequence errors or polymorphisms that are shared by only a few individuals. This distribution usually follows the asymptotic relationship seen in the figure below:
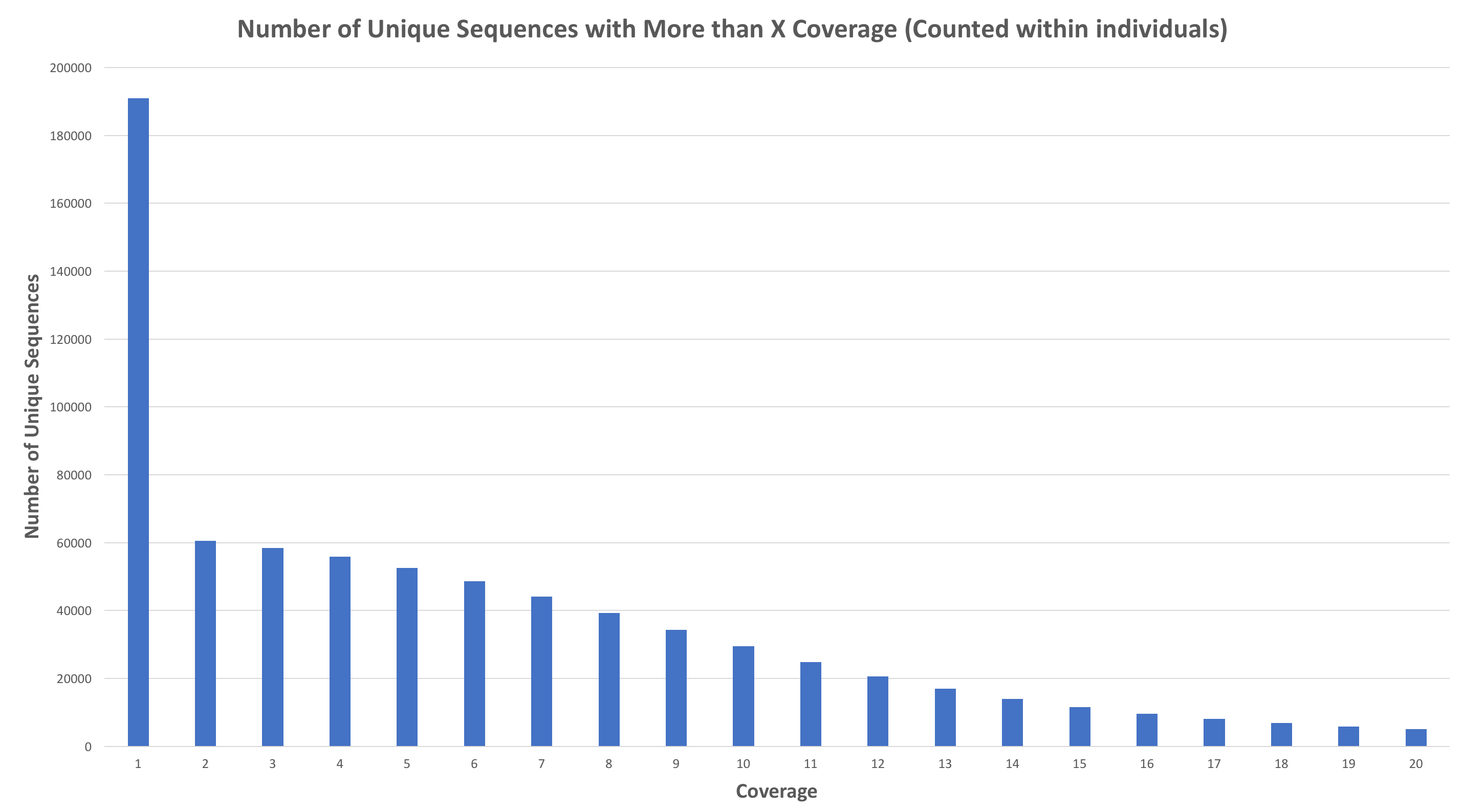
Where there are a large proportion of reads that only have one or two occurrences, meaning they will not likely be informative on the population scale. Even if a locus was polymorphic to the point that it was unique to the individual, one can expect that one unique allele would be present at least the level of coverage expected from the sequencing strategy, for example 10X or 20X coverage. This is where we expect to see the slope of the distribution even out. Experimentation for the best value will be needed for each taxa. Set the cutoff too low, and extraneous reads will be included further down the pipeline and eat up valuable computational time and make it more likely that sequencing errors are included in the data. Set the cutoff too high, and usable polymorphic loci may be excluded from subsequent analyses.
After the first cutoff is chosen, dDocent then tabulates the remaining unique reads across individuals. The user again can input a cutoff value removing unique reads that appear in only a few individuals. To illustrate the utility of this take a look at the haplotype networks from a simulated RAD locus.
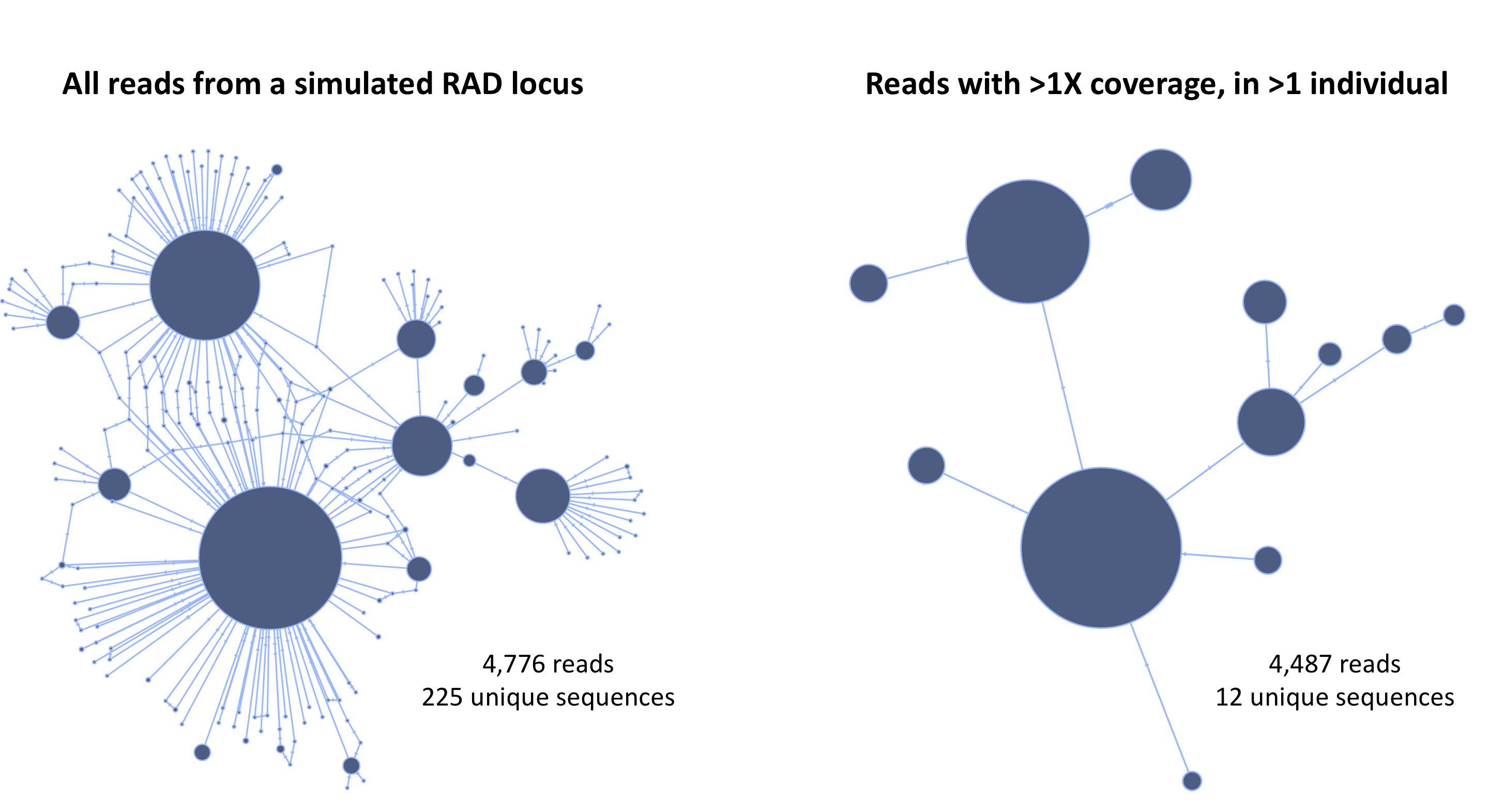
Using the two cutoffs greatly simplifies the data and allows for much more accurate assembly.
PE and RPE methods
After data cutoffs are chosen, the PE and RPE methods go through a hybrid assembly method that combines the utility of alignment-based read clustering via CD-HIT with the RAD specific assembly program Rainbow. First, reads are clustered using only the forward reads with CD-HIT and dDocent converts the output to be ported directly into Rainbow. Rainbow recursively divids clusters based on reverse reads into groups representing single alleles. Reads in merged contigs are then assembled using a greedy algorithm. dDocent then chooses the most frequent or longest contig as the representative reference sequence for that contig. If the forward read does not overlap with the reverse read (almost always the case with ddRAD), the forward read is pasted to the reverse read with a ten N basepairs as padding. dDocent then ports the forward and reverse parts of each contig into the program PEAR to again check for substantial overal between forward and reverse reads. Finally, reference sequences are clustered based on overall sequence similarity using CD-HIT.
OL method
For the OL method, the reduced data set is input into PEAR to merge overlapping forward and reverse reads. Merged reads are then clustered based on overall sequence similarity using CD-HIT.
SE method
For the SE method, the reduced data set is clustered based on overall sequence similarity using CD-HIT.
User supplied reference
Alternatively, de novo assembly can be skipped and the user can provide a fasta file with reference sequences. This file needs to be simply named reference.fasta
Read Mapping
Reads are mapped (aligned to reference sequences) using the MEM algorithm of BWA. The user can choose to set alternative values for the match score value, mismatch score, and gap opening penalty. According the the BWA manual, the sequence error rate is approximately: {.75 * exp[-log(4) * B/A]}. Again, experimentation for finding the optimal values for a particular taxa is recommended. The default settings are meant for mapping reads to the human genome and are fairly conservative. BWA outputs SAM files which are then converted to BAM files using SAMtools.
SNP Calling
dDocent uses a scatter gather technique to speed up SNP/INDEL calling. In short, reference contigs are split up into a single file for each processing core. FreeBayes then calls variants for each genomic interval simultaneously (changes to default parameters include setting a minimum mapping and base quality score to PHRED 10). FreeBayes is a Bayesian-based variant detection program that uses assembled haplotype sequences to simultaneously call SNPs, INDELS, multi-nucleotide polymorphisms (MNPs), and complex events (composite insertion and substitution events) from alignment files; FreeBayes has the added benefit for population genomics of using reads across multiple individuals to improve genotyping. After all instances of FreeBayes finish, raw SNP/INDEL calls are concatenated into a single variant call file (VCF) using VCFtools.
SNP Filtering
Final SNP data sets will depend on the individual project, so dDocent does only minimal filtering. Using VCFtools, SNPs are filtered to only those that are called in 90% of all individuals. They can be found in Final.recode.vcf; however, these are mainly for diagnostic purposes between runs. VCFtools can be used to filter SNP and INDEL calls using a variety of criteria and it is recommended that users familiarize themselves with the program to produce a truly final call set.
The dDocent package includes various scripts for more advanced SNP filtering. A thorough tutorial can be found in SNP Filtering Tutorial.
Getting Started
This section of the user guide will walk you through using the pipeline.
Requirements
After you have dDocent properly installed (see Bioconda Install and Manual Install), you need to create a working directory:
mkdir my_dDocent_working_dir
Raw Sequences
In this directory, you need to place RAW, DEMULTIPLEXED sequencing files. In other words, one raw gzipped fastq file (or sets of files for paired sequencing) for each individual.
If you are using process_radtags to demultiplex, avoid using the -c and -q parameters.
Trimmed reads will fail de novo assembly
If performing de novo assembly, it’s essential that no read trimming or adapter removal has taken place before the dDocent pipeline. If a reference is being supplied, then trimmed reads may be used.
Naming Convention
dDocent requires that sequence files be gzipped FASTQ format and the files MUST MUST MUST follow a specific naming convention.File names must contain a locality/population identifier and an individual identifier, and these two identifiers must be separated by a single _
For example:
Pop1_Sample1.F.fq.gz Pop1_Sample1.R.fq.gz
Please don’t use _ in either of the identifiers.
dDocent uses raw reads for reference assembly and trimmed reads for read mapping and SNP/variant calling. If dDocent is not being used for trimming, trimmed reads must already be in the directory and must follow the naming convention below:
Pop1_001.R1.fq.gz Pop1_001.R2.fq.gz
Pop1_002.R1.fq.gz Pop1_002.R2.fq.gz
Where R1 are trimmed forward reads and R2 are trimmed paired-end reads (If PE sequencing is being used).
Running
Simply type:
dDocent
Now, dDocent will start interactive configuration by asking you questions:
dDocent 2.2.7
Contact jpuritz@gmail.com with any problems
Checking for required software
All required software is installed!
dDocent run started Sat Dec 17 23:28:22 EST 2016
40 individuals are detected. Is this correct? Enter yes or no and press [ENTER]
To start, dDocent is checking for all the required software and then counting the number of individuals that it finds in the current directory. This is just a basic check to make sure you’re in the correct directory. Simple enter yes to continue
dDocent detects 72 processors available on this system.
Please enter the maximum number of processors to use for this analysis.
Next, dDocent tries to automatically detect the number of processors available on your system. It then lets you set a hard limit for the number of processors to use simultaneously during a dDocent run. Simply enter the number you want to continue. Let’s say 32 for this example.
dDocent detects 251G maximum memory available on this system.
Please enter the maximum memory to use for this analysis. The size can be postfixed with
K, M, G, T, P, k, m, g, t, or p which would multiply the size with 1024, 1048576, 1073741824,
1099511627776, 1125899906842624, 1000, 1000000, 1000000000, 1000000000000, or 1000000000000000 respectively.
For example, to limit dDocent to ten gigabytes, enter 10G or 10g
dDocent tries to automatically detect the maximum amount of memory on your system. It then lets you set a hard limit for the amount of memory that it will use. This will only apply to SNP calling.
This option does not work on all systems!!!!!
GNU-Parallel implements this option and it unfortunately does not work properly on all LINUX distributions, and any value other than 0 may cause SNP calling to hold indefinitely. Unless this has ben tested on your system, I recommend entering:
0
Next dDocent asks about trimming:
Do you want to quality trim your reads?
Type yes or no and press [ENTER]?
If this is the first time running dDocent on the data set, the raw sequence files MUST be trimmed. However, trimming only needs to be performed once. Let’s assume the answer is yes for this example.
Next dDocent asks if you want to assemble your data:
Do you want to perform an assembly?
Type yes or no and press [ENTER].
If you are supplying your own reference file or dDocent has already been used to create an assembly, then answer no to this question. For this example, let’s answer yes.
What type of assembly would you like to perform? Enter SE for single end, PE for paired-end, RPE for paired-end sequencing for RAD protocols with random shearing, or OL for paired-end sequencing that has substantial overlap.
Then press [ENTER]
Please see the De novo Assembly section above for an explanation of the different choices. Let’s use PE for this walkthrough.
PE
Reads will be assembled with Rainbow
CD-HIT will cluster reference sequences by similarity. The -c parameter (% similarity to cluster) may need to be changed for your taxa.
Would you like to enter a new c parameter now? Type yes or no and press [ENTER]
dDocent is asking for a percent similarity to cluster reads by, this number will be used in the final contig clustering using CD-HIT, and for the PE and RPE method, initial clustering of forward reads will be set to either 0.8 or c parameter mins 0.1 which ever is higher. Let’s use 0.85.
Would you like to enter a new c parameter now? Type yes or no and press [ENTER]
yes
Please enter new value for c. Enter in decimal form (For 90%, enter 0.9)
0.85
Next dDocent will ask if you want to map reads:
Do you want to map reads? Type yes or no and press [ENTER]
Read mapping needs to be performed at least once before SNP calling, but does not need to be repeated if the reference and read trimming remains the same. Assuming this is the first time running dDocent, let’s answer yes.
dDocent will now ask if you want to change some read mapping parameters. Let’s say yes.
BWA will be used to map reads. You may need to adjust -A -B and -O parameters for your taxa.
Would you like to enter a new parameters now? Type yes or no and press [ENTER]
yes
Please enter new value for A (match score). It should be an integer. Default is 1.
The first parameter is the match score. See Read Mapping above to see how this parameter is used in mapping.
Please enter new value for A (match score). It should be an integer. Default is 1.
1
Please enter new value for B (mismatch score). It should be an integer. Default is 4.
The second parameter is the mismatch score (penalty). See Read Mapping above to see how this parameter is used in mapping. Let’s use the default, 4.
Please enter new value for A (match score). It should be an integer. Default is 1.
1
Please enter new value for B (mismatch score). It should be an integer. Default is 4.
4
Please enter new value for O (gap penalty). It should be an integer. Default is 6.
The third parameter is the gap opening penalty. See Read Mapping above to see how this parameter is used in mapping. Let’s use the default, 6.
Do you want to use FreeBayes to call SNPs? Please type yes or no and press [ENTER]
Unless you are using dDocent for some custom applications, this will likely always be yes
Lastly, dDocent will ask for an email address to send a notification when it’s finished running.
Please enter your email address. dDocent will email you when it is finished running.
Don't worry; dDocent has no financial need to sell your email address to spammers.
This will only work on systems with mailx installed and with open internet connections, so it may not work on your system out of the box.
This completes the configuration portion of dDocent and the program begins to run. You will immediately see this prompt:
dDocent will require input during the assembly stage. Please wait until prompt says it is safe to move program to the background.
Trimming reads and simultaneously assembling reference sequences
After a short time, dDocent will prompt you for input:
Number of Unique Sequences with More than X Coverage (Counted within individuals)
70000 +-+---------+-----------+-----------+-----------+-----------+-----------+-----------+-----------+---------+-+
+ + + + + + + + + +
| |
60000 ****** +-+
| ****** |
| ****** |
| ****** |
50000 +-+ ***** +-+
| * |
| ****** |
40000 +-+ ***** +-+
| * |
| ****** |
30000 +-+ ***** +-+
| * |
| ****** |
20000 +-+ ****** +-+
| ****** |
| ****** |
| ****** |
10000 +-+ ************ +-+
| *************
+ + + + + + + + + +
0 +-+---------+-----------+-----------+-----------+-----------+-----------+-----------+-----------+---------+-+
2 4 6 8 10 12 14 16 18 20
Coverage
Please choose data cutoff. In essence, you are picking a minimum (within individual) coverage level for a read (allele) to be used in the reference assembly
This is generated from simulated data, so it may not look like your data. You need to choose a cutoff value. You want to choose a value that captures as much of the diversity of the data as possible while simultaneously eliminating sequences that are likely errors. Typically, you’re looking for some sort of inflection in the curve. Let’s try 6. Shortly, a new prompt appears:
Number of Unique Sequences present in more than X Individuals
5500 +-+---------+-----------+-----------+-----------+------------+-----------+-----------+-----------+---------+-+
** + + + + + + + + +
5000 +-* +-+
| ** |
| * |
4500 +-+ +-+
| *** |
4000 +-+ ** +-+
| * |
3500 +-+ ***** +-+
| * |
3000 +-+ ***** +-+
| * |
| ****** |
2500 +-+ ****** +-+
| ***** |
2000 +-+ * +-+
| ************* |
1500 +-+ ****** +-+
| ************ |
| ************ |
1000 +-+ ************ +-+
+ + + + + + + + *************
500 +-+---------+-----------+-----------+-----------+------------+-----------+-----------+-----------+---------+-+
2 4 6 8 10 12 14 16 18 20
Number of Individuals
Please choose data cutoff. Pick point right before the asymptote. A good starting cutoff might be 10% of the total number of individuals
This is from a simulated data set with 40 individuals, and a cutoff of 4 lines up well with the inflection in the curve. Let’s enter 4.
dDocent now outputs:
At this point, all configuration information has been entered and dDocent may take several hours to run.
It is recommended that you move this script to a background operation and disable terminal input and output.
All data and logfiles will still be recorded.
To do this:
Press control and Z simultaneously
Type 'bg' without the quotes and press enter
Type 'disown -h' again without the quotes and press enter
Follow the instructions and then simply wait for dDocent to finish.
Running with configuration file
dDocent can also be run in non-interactive mode and configuration parameters can be entered with a configuration file. The file can be named anything, but must follow the EXACT format below:
Number of Processors
24
Maximum Memory
0
Trimming
no
Assembly?
yes
Type_of_Assembly
PE
Clustering_Similarity%
0.86
Minimum within individual coverage level to include a read for assembly (K1)
2
Minimum number of individuals a read must be present in to include for assembly (K2)
2
Mapping_Reads?
yes
Mapping_Match_Value
1
Mapping_MisMatch_Value
3
Mapping_GapOpen_Penalty
5
Calling_SNPs?
yes
Email
jpuritz@gmail.com
Now run dDocent and pass the configuration file:
dDocent config.file
Outputs
dDocent will output several different files as part of the pipeline.
Data outputs
TotalRawSNPs.vcf
This file, in the standard Variant Call Format, has the raw SNP, INDEL, MNP, and complex variant calls for every individual. This is the file that will be used for further filtering (see VCFtools or vcflib) to produce the final data set. It is important to note that FreeBayes combines SNP and INDEL calls that are in within a default 3bp window into haplotype calls of the complex variant calls. To properly look at SNPs only, complex variants need to be decomposed with vcfallelicprimatives from the vcflib package and then INDELs can be filtered with VCFtools or vcflib. See the SNP Filtering Tutorial for more information on SNP filtering.
Final.recode.vcf
This is the filtered VCF file from the very end of the pipeline. It’s useful for comparing different runs of the pipeline.
X.bam
These files are Binary Alignment Maps (BAMs). They are the mapping of reads for every individual to the reference contigs. BAM files are input directly into FreeBayes for variant calling. Note, this file cannot be viewed directly; use SAMtools for inspection.
reference.fasta
These are the contigs generated by the de novo assembly in FASTA format.
uniqseq.data
This file is a tab delimited file the level of coverage in the first column and the number of unique sequences with more than that level of coverage is the second column.
Log files
dDocent_main.LOG
This file captures all terminal output during the dDocent run and will be your goto file if you encounter any problems with dDocent
dDocent.runs
This file captures simply the run configuration parameters for every dDocent run. The file can be easily modified into a dDocent configuration file. See Running with a configuration file.
logfiles directory
dDocent creates a directory called logfiles and will store various other logfiles there. These are generally only necessary for debugging purposes.
Temporary Files
dDocent creates a variety of temporary files during a run. Below is a list and a brief description for each:
| File Name | Description |
|---|---|
| bamlist.list | a list of all BAM files passed to freebayes |
| contig.cluster.totaluniqseq | used during assembly |
| cov.stats | file in .bed format that contains overall levels of coverage across reference contigs |
| Final.frq.count | output from VCFtools |
| mapped.bed | this file contains all intervals used for SNP calling |
| namelist | a list of all sample names |
| other.FR | used during assembly |
| overlap.* | files used during the PEAR portion of assembly |
| popmap | tab delimited files that designates a population for each sample based on file name. Used during SNP calling |
| rainbow.fasta.gz | gzipped temp file from Rainbow |
| raw.vcf | a directory containing individual VCF files from SNP scatter gather which are latter combined into TotalRawSNPs.vcf |
| rbasm.out.gz | gzipped temp file from Rainbow |
| rbdiv.out.gz | gzipped temp file from Rainbow |
| rcluster.gz | gzipped temp file from Rainbow |
| reference.fasta.amb | index file for reference |
| reference.fasta.ann | index file for reference |
| reference.fasta.bwt | index file for reference |
| reference.fasta.fai | index file for reference |
| reference.fasta.original.clstr | temp from assembly |
| reference.fasta.original.gz | temp from assembly |
| reference.fasta.pac | index file for reference |
| reference.fasta.sa | index file for reference |
| sort.contig.cluster.ids | file used for conversion of CD-HIT output to Rainbow input |
| totalover.fasta | created during reference assembly |
| totaluniqseq.gz | all unique sequences remaining after data cutoffs and adapter trimming |
| uniqCperindv | unique sequences after cutoff across individuals |
| uniq.fasta.gz | gzipped FASTA format file of totaluniqseq.gz |
| uniq.F.fasta.gz | gzipped FASTA format file of only first reads from totaluniqseq.gz |
| uniq.full.fasta | all unique sequences remaining after data cutoffs in FASTA format |
| uniq.k.6.c.4.seqs | unique sequences and counts after data cutoffs |
| uniqseq.data | coverage counts for unique sequences |
| uniqseq.peri.data | counts for unique sequences across individuals |
| uniq.seqs.gz | all unique sequences |
| unpaired | directory containing sequences that failed during read trimming and were removed |
| xxx.* | files used during read clustering during assembly |
For information on the other outputs, refer to the code comments in the dDocent file.
Customizing dDocent
Further customization of the pipeline can be achieved by modifying the script directly. Any text editor can be used. Below is a guide on how to modify specific sections of code:
Read Trimming Customization
By default, dDocent looks for Illumina TruSeq adapters and trims off basepairs with a quality score less than 20. Both BWA and FreeBayes take base quality scores into account, so excessive trimming is not necessary nor recommended. To modify this find line 458.
Visit the Trimmomatic website for more information on settings.
Assembly Customization
Settings for Rainbow can be modified on lines 629 and 630. Please see the Rainbow documentation for more information.
Read Mapping Customization
Through the command line interface, dDocent users already are able to customize several read mapping parameters for BWA. BWA does have additional options that can be added to line 267 for PE data with a dDocent made reference, line [269 for SE data with a dDocent made reference, line [279for PE data with a user supplied reference, and line [281 for SE data with a user supplied reference.
If using dDocent for something other than RADseq, it may be advisable to remove the high clipping penalties (-L parameter) in all lines above as well as the mawk code that removes heavily clipped reads (mawk '$6 !~/[2-9].[SH]/ && $6 !~ /[1-9][0-9].[SH]/' ).
SNP Calling Customization
FreeBayes is a highly customizable variant calling program and can be adapted to many different needs. To see all the different possible options of FreeBayes, simply type:
freebayes -h
This brings up the FreeBayes help menu that lists and explains all options. Options that may be of interest are listed below:
--populations FILE
If you are expecting your data to be highly structured, you may consider using this option. FreeBayes does do some population specific calculations for heterozygosity levels and mutation rates. This allows you to partition the population model. To do this, you must supply a file that lists the sample and it’s population. Note, you should only use this is you have a strong a priori reason to expect substantial population structure.
-E
This parameter affects how haplotypes are called in the VCF file. The default value is 3, meaning that variants within 3 bp of each other will be called as a single contiguous haplotype. This represents the highest tradeoff between haplotype length and sensitivity. See this discussion for more information.
-m
This parameter controls the minimum mapping quality score for reads to be considered for genotyping. By default, dDocent sets this to PHRED 10 (or 90% probability of being true).
-q
This sets the minimum base quality score for a bp to be used for genotyping. By default, dDocent sets this to PHRED 10 (or 90% probability of being true).
-V
This parameter tells FreeBayes to ignore read placement bias and strand bias in the calculation of SNP site quality scores. By default, dDocent leaves this parameter in because it, at worst, conservatively biases quality scores. However, this may be too conservative because one would expect extreme strand bias in RAD sequencing.
To customize FreeBayes these parameters can be changed or altered on line 355
VCF Filtering Customization
To customize the basic VCF filtering performed by dDocent, simply edit line 387. Please see the VCFtools documentation for a list of options.